Cicloaddizione 1,3-dipolare
Una cicloaddizione 1,3-dipolare è una reazione chimica tra un dipolo 1,3 e un composto detto dipolarofilo per formare un anello a cinque termini. Descritto per la prima volta nel 1889 da Buchner,[1] il meccanismo di queste cicloaddizioni fu chiarito solamente negli anni 1960, principalmente grazie al lavoro di Rolf Huisgen,[2] motivo per cui sono spesso definite cicloaddizioni di Huisgen (quando sono coinvolti un'azide e un alchino).
Le cicloaddizioni 1,3-dipolari manifestano notevole regioselettività e stereoselettività e rientrano nel novero delle reazioni pericicliche (sono delle cicloaddizioni [4+2]). Si ottengono eterocicli a 5 membri, aromatici nel caso si impieghi un alchino come dipolarofilo.

Panoramica sul meccanismo modifica
Inizialmente si dibatté a lungo sulla natura di queste cicloaddizioni. L'ipotesi più accreditata prevedeva un meccanismo periciclico, proposto da Huisgen,[3] ma vi erano anche sostenitori di un processo a più stadi che coinvolgeva un intermedio diradicalico (come suggerito da Firestone).[4] Dopo un decennio di discussioni e grazie agli avanzamenti tecnologici, nel 1976 fu possibile confermare la periciclicità delle reazioni.[5]
Il dipolo 1,3 reagisce con il dipolarofilo con un meccanismo concertato, spesso asincrono, rappresentando una cicloaddizione [π4s + π2s], passando attraverso uno stato di transizione ciclico aromatico secondo Hückel. Vi sono, tuttavia, due eccezioni, rappresentate dalle reazioni delle ilidi di tiocarbonili[6] e di ossidi di nitrili.[7]

Meccanismo periciclico modifica
Huisgen investigò una serie di cicloaddizioni tra i composti 1,3-dipolari azotati e vari alcheni dipolarofili.[3] Le osservazioni di seguito riportate sono a sostegno del meccanismo periciclico e confutano le ipotesi di un decorso a stadi.
- Effetto dei sostituenti. Sostituendi diversi sul dipolo non alterano significativamente la velocità della cicloaddizione, suggerendo che il processo non passa attraverso un intermedio con cariche separate.
- Effetto dei solventi. La polarità del solvente non influenza la velocità della reazione, in linea con quanto atteso dalle reazioni pericicliche.
- Stereochimica. Le reazioni 1,3-dipolari sono sempre stereospecifiche rispetto al dipolarofilo (es. un alchene cis darà sempre un prodotto di addizione sin), in linea con il principio delle reazioni pericicliche che formano due nuovi legami sigma simultaneamente.
- Termodinamica. Queste reazioni hanno un'inusuale entropia di attivazione molto negativa, similmente a quanto riscontrato per la reazione di Diels-Alder, anch'essa periciclica e cicloaddizione.
Dipolo 1,3 modifica

Un composto dipolare 1,3 è una molecola organica rappresentabile sia come una struttura allilica sia come uno zwitterione propargile-allenile.[8] Esistono anche dipoli contenenti zolfo o fosforo, ma sono raramente impiegati.
Si possono disegnare più strutture di risonanza che delocalizzino le cariche positive e negative su un qualsiasi atomo, ma in genere per una rappresentazione corretta ci si affida a calcoli sperimentali o computazionali.[9][10] Un esempio di 1,3-dipolo classico è il diazometano, come mostrato nella figura sottostante.

Per via della sua ambivalenza, le estremità di un composto 1,3-dipolare possono comportarsi sia da nucleofilo che da elettrofilo. Il comportamento più o meno di un tipo si può valutare ricorrendo alla teoria degli orbitali molecolari di frontiera, dunque con calcoli computazionali.
In genere si può dire che l'atomo che esibisce il coefficiente orbitalico maggiore nell'HOMO sarà il nucleofilo, mentre nel LUMO sarà l'elettrofilo. Spesso e volentieri, ma non necessariamente, l'atomo più elettronricco si comporta da nucleofilo.[11][12][13]
Dipolarofilo modifica
I dipolarofili più comuni sono alcheni e alchini. I dipolarofili contenenti un eteroatomo, come nel caso dei carbonili e delle immine, possono subire una cicloaddizione 1,3-dipolare. Altri esempi di dipolarofili includono i fullereni e i nanotubi nella cosiddetta reazione di Prato.
Effetto dei solventi modifica
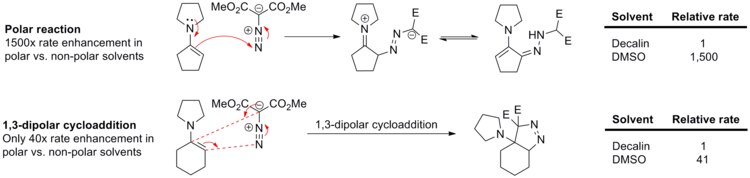
Come già anticipato, queste reazioni risentono poco e nulla di un effetto da parte del solvente, poiché sia i reagenti che gli stati di transizione non sono polari. Di seguito è riportato un esempio di come è influenzata la costante cinetica nel caso della reazione tra il fenil-diazometano e l'etil acrilato oppure il norbornene, passando da cicloesano a metanolo.[14]

L'assenza di una qualche influenza da parte del solvente si dimostra incontrovertibilmente osservando la reazione delle enammine con il dimetil diazomalonato.[15] La reazione polare nello schema di sotto viene accelerata di 1500 volte, mentre la cicloaddizione 1,3-dipolare viene velocizzata di appena 40 volte, quindi l'effetto è assai meno pronunciato.
Teoria degli orbitali molecolari di frontiera modifica
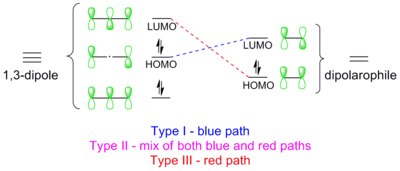
Le reazioni 1,3-dipolari sono delle reazioni periciliche che obbediscono alle regole di Dewar-Zimmerman e alle equivalenti regole di Woodward-Hofmann. Nell'esempio riportato qui sopra, nella prima interpretazione, la reazione procede attraverso uno stato di transizione aromatico secondo Huckel. Nella seconda interpretazione, gli orbitali di frontiera del dipolo e del dipolarofilo si sovrappongono in modo permesso dalle regole di simmetria.
Poiché vi sono più modi con cui la reazione può evolvere, si è soliti fare una distinzione in tre tipi diversi di cicloaddizione 1,3-dipolare, in base al modo in cui gli orbitali si sovrappongono.[16] La tipologia predominante prevede un'interazione HOMO-LUMO con il minore gap energetico possibile.
Tipo I modifica
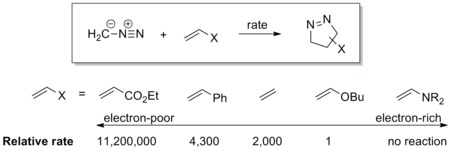
Il dipolo ha un HOMO energetico che si sovrappone al LUMO del dipolarofilo. Un dipolo di questo tipo è definito dipolo sotto controllo dell'HOMO oppure dipolo nucleofilo. Esempi includono delle ilidi azotate e i diazocomposti. Questi dipoli si addizionano agli alcheni elettrofili rapidamente. I gruppi elettronattrattori (electron withdrawing group, EWG) sul dipolarofilo possono accelerare la reazione abbassando l'energia del LUMO, mentre i gruppi elettrondonatori (electron donating group, EDG) la rallentano perché alzano l'energia dell'HOMO.
Ad esempio, la scala di reattività del diazometano, nei confronti di una serie di dipolarofili, è mostrata in basso. Il diazometano reagisce con l'etil acrilato elettronpovero oltre un milione di volte più velocemente di quanto non faccia con il butil vinil etere, elettronricco.[17]
Questo tipo di cicloaddizione ricorda la reazione di Diels-Alder a normale domanda elettronica, in cui l'HOMO del diene si combina con il LUMO del dienofilo.
Tipo II modifica
L'HOMO del dipolo si accoppia con il LUMO del dipolarofilo. In alternativa l'HOMO del dipolarofilo si accoppia con il LUMO del dipolo. Questa interazione in due sensi si origina dal fatto che il salto energetico tra i due orbitali di frontiera (in un senso o nell'altro) è molto simile (e facilmente sormontabile). Un dipolo di questa categoria viene definito dipolo sotto controllo HOMO-LUMO oppure dipolo anfifilico. Alcuni esempi sono i le immidi di nitrili, i nitroni, i nitril-ossidi e le azidi.
Un sostituente qualsiasi sul dipolarofilo può accelerare la reazione riducendo il gap tra i due orbitali interagenti. Un EWG ridurrebbe l'energia del LUMO, mentre un EDG aumenterebbe quella dell'HOMO. A titolo esemplificativo è riportata la scala di reattività delle azidi nei confronti di diversi composti dipolarofili elettronpoveri.[18]

Tipo III modifica
Il dipolo ha un LUMO a bassa energia che si sovrappone all'HOMO del dipolarofilo (mostrato con delle linee tratteggiate rosse nel diagramma a inizio sezione). Un dipolo del genere è noto come dipolo sotto controllo del LUMO oppure dipolo elettrofilo, come l'ossido di diazoto e l'ozono. Gli EWG sul dipolarofilo decelerano la reazione, mentre gli EDG la accelerano. Ad esempio l'ozono reagisce con il 2-metilpropene (elettronricco) circa centomila volte più rapidamente che con l'elettronpovero tetracloroetene.[19]
Questo tipo di reazione ricorda la reazione di Diels-Alder a domanda elettronica inversa, in cui il LUMO del diene si combina con l'HOMO del dienofilo.

Reattività modifica
I processi concertati come le cicloaddizioni 1,3-dipolari richiedono uno stato di transizione molto ordinato (entropia di attivazione molto negativa e moderate entalpie). Alcuni fattori che influenzano la reattività sono elencati di seguitoː
- Coniugazioneː specialmente con i gruppi aromatici, comporta un aumento della velocità di reazione grazie alla stabilizzazione extra per risonanza. Durante la transizione, due legami sigma si formano a velocità diverse, generando delle cariche parziali nello stato di transizione che, però, possono essere stabilizzate grazie a dei sostituenti.
- L'ingombro sterico nello stato di transizione diminuisce la velocità di reazione e può anche ostacolarla.
- I dipolarofili più polarizzabili sono più reattivi in quanto la nube elettronica è maggiormente predisposta a muoversi tra due molecole.
- L'isomeria del dipolarofilo influisce sulla velocità di reazione per motivi sterici. Gli isomeri trans, infatti, sono più reattivi perché meno ingombri e meno tensionati a livello di angoli di legame. Per esempio il trans-stilbene si addiziona alla difenil-nitril immide 27 volte più velocemente del cis-stilbene.

See Huisgen reference DOI: 10.1002/anie.196306331.

Stereospecificità modifica
Le cicloaddizioni 1,3-dipolari di solito comportano una ritenzione di configurazione rispetto sia al dipolo che al dipolarofilo. Questa elevata stereospecificità è a sostegno del meccanismo periciclico.
Stereospecificità rispetto al dipolarofilo modifica
I sostituenti cis sull'alchene dipolarofilo portano ad un prodotto cis, mentre i sostituenti trans restano trans nel ciclo a 5 atomi che si ottiene dalla reazione.[20]

Stereospecificità rispetto al dipolo modifica
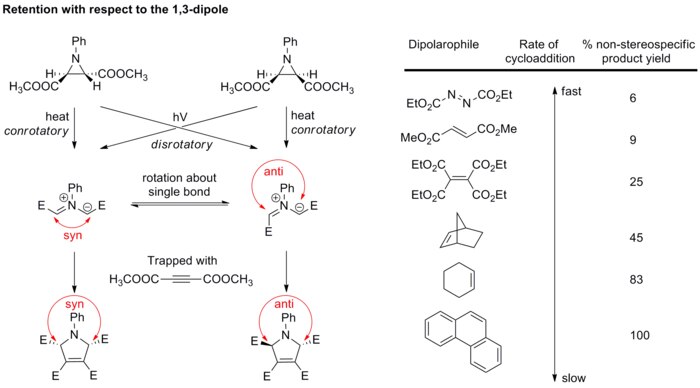
Generalmente, la stereochimica del dipolo non è molto tenuta in considerazione perché solo pochi dipoli sono in grado di formare dei centri stereogenici. Comunque si ha un'influenza da parte dei sostituenti anche in questo caso. Un esempio è riportato nella figura sottostante.[21][22]
Regioselettività modifica
Per coppie dipolo-dipolarofilo asimmetriche, si possono ottenere due regioisomeri. Fattori sia sterici che elettronici vanno invocati per spiegare il decorso delle reazioni.[23]
Effetti elettronici modifica
L'interazione elettronica dominante è la combinazione tra l'HOMO più grande e il LUMO più grande. La regioselettività, quindi, è governata da quegli atomi che recano su di sé i coefficienti HOMO e LUMO maggiori.[24][25]
Si consideri la cicloaddizione del diazometano a tre diversi dipolarofiliːmetil acrilato, stirene e metil cinnamato. Il carbonio del diazometano reca l'HOMO più largo, mentre l'estremità olefinica del metil acrilato e dello stirene ha il LUMO più alto. Pertanto, la cicloaddizione porta alla sostituzione in posizione 3, regioselettivamente. Per il metil cinnamato, i due sostituenti (il fenile e il gruppo estereo) competono per attirare su di sé gli elettroni dell'alchene. Il carbossile è un EWG migliore del fenile e questo rende il carbonio in posizione beta più elettrofilo. Ecco dunque che la cicloaddizione porterà alla sostituzione del carbossile in posizione 3 e del fenile in posizione 4.

Effetto sterico modifica
Gli effetti sterici possono favorire o sfavorire gli effetti elettronici sopracitati. A volte addirittura portano all'altro regioisomero.[3]
Per esempio, il diazometano generalmente si addiziona al metil acrilato per dare la 3-carbossil pirazolina. Ad ogni modo, aumentando la domanda sterica del sistema, ci si accorge che si forma l'isomero 4-carbossil pirazolina. Il rapporto tra i due regioisomeri dipende dalla domanda sterica.[26]

Applicazioni sintetiche modifica
Ozono modifica
La reazione di ozonolisi è un esempio di cicloaddizione 1,3-dipolare, entro certi limiti. Con questo meccanismo, a partire da alcheni o alchini si ottengono aldeidi, chetoni o acidi carbossilici.
Note modifica
- ^ Mattia Giacomello, Cicloaddizione 1,3-dipolare di un diazoalcano coordinato con alcheni e alchini, su dspace.unive.it. URL consultato il 3 settembre 2020.
- ^ Rolf Huisgen, 1.3-Dipolare Cycloadditionen Ruckschau und Ausblick., in Angewandte Chemie, vol. 75, n. 13, 1963, pp. 604–637, DOI:10.1002/ange.19630751304.
- ^ a b c Rolf Huisgen, Kinetics and Mechanism of 1,3-Dipolar Cycloadditions, in Angewandte Chemie International Edition, vol. 2, n. 11, novembre 1963, pp. 633–645, DOI:10.1002/anie.196306331.
- ^ R Firestone, Mechanism of 1,3-dipolar cycloadditions, in Journal of Organic Chemistry, vol. 33, n. 6, 1968, pp. 2285–2290, DOI:10.1021/jo01270a023.
- ^ Rolf Huisgen, 1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates, in Journal of Organic Chemistry, vol. 41, n. 3, 1976, pp. 403–419, DOI:10.1021/jo00865a001.
- ^ G. Mloston, E. Langhals e Rolf Huisgen, First Two-Step 1,2-Dipolar Cycloadditons: Nonstereospecificity, in J. Am. Chem. Soc., vol. 108, n. 20, 1986, pp. 6401–66402, DOI:10.1021/ja00280a053.
- ^ Siadati Seyyed Amir, An example of a stepwise mechanism for the catalyst-free 1,3-dipolar cycloaddition between a nitrile oxide and an electron rich alkene, in Tetrahedron Letters, vol. 56, n. 34, 2015, pp. 4857–4863, DOI:10.1016/j.tetlet.2015.06.048.
- ^ Rolf Huisgen, 1,3-Dipolar Cycloadditions. Past and Future, in Angewandte Chemie International Edition, vol. 2, n. 10, 1963, pp. 565–598, DOI:10.1002/anie.196305651.
- ^ A Cox, L Thomas e J Sheridan, Microwave Spectra of Diazomethane and its Deutero Derivatives, in Nature, vol. 181, n. 4614, 1958, pp. 1000–1001, Bibcode:1958Natur.181.1000C, DOI:10.1038/1811000a0.
- ^ P Hilberty e C Leforestier, Expansion of molecular orbital wave functions into valence bond wave functions. A simplified procedure., in Journal of the American Chemical Society, vol. 100, n. 7, 1978, pp. 2012–2017, DOI:10.1021/ja00475a007.
- ^ J.F. McGarrity e Saul Patai, Basicity, acidity and hydrogen bonding, in Diazonium and Diazo Groups, vol. 1, 1978, pp. 179–230, DOI:10.1002/9780470771549.ch6, ISBN 978-0-470-77154-9.
- ^ Daniel Berner e John McGarrity, Direct observation of the methyldiazonium ion in fluorosulfuric acid, in Journal of the American Chemical Society, vol. 101, n. 11, 1979, pp. 3135–3136, DOI:10.1021/ja00505a059.
- ^ Eugen Muller e Wolfgans Rundel, Untersuchungen an Diazomethanen, VI. Mitteil.: Umsetzung von Diazoäthan mit Methyllithium, in Chemische Berichte, vol. 89, n. 4, 1956, pp. 1065–1071, DOI:10.1002/cber.19560890436.
- ^ Jochen Geittner, Rolph Huisgen e Hans-Ulrich Reissig, Solvent Dependence of Cycloaddition Rates of Phenyldiazomethane and Activation Parameters, in Heterocycles, vol. 11, 1978, pp. 109–120, DOI:10.3987/S(N)-1978-01-0109.
- ^ Rolph Huisgen, Hans-Ulrich Reissig, Helmut Huber e Sabine Voss, α-Diazocarbonyl compounds and enamines - a dichotomy of reaction paths, in Tetrahedron Letters, vol. 20, n. 32, 1979, pp. 2987–2990, DOI:10.1016/S0040-4039(00)70991-9.
- ^ R Sustmann, Orbital energy control of cycloaddition reactivity, in Pure and Applied Chemistry, vol. 40, n. 4, 1974, pp. 569–593, DOI:10.1351/pac197440040569.
- ^ Jochen Geittner e Rolf Huisgen, Kinetics of 1,3-dipolar cycloaddition reactions of diazomethane; A correlation with homo-lumo energies, in Tetrahedron Letters, vol. 18, n. 10, 1977, pp. 881–884, DOI:10.1016/S0040-4039(01)92781-9.
- ^ Rolf Huisgen, Gunter Szeimies e Leander Mobius, K1.3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen, in Chemische Berichte, vol. 100, n. 8, 1967, pp. 2494–2507, DOI:10.1002/cber.19671000806.
- ^ D. G. Williamson e R. J. Cvetanovic, Rates of ozone-olefin reactions in carbon tetrachloride solutions, in Journal of the American Chemical Society, vol. 90, n. 14, 1968, pp. 3668–3672, DOI:10.1021/ja01016a011.
- ^ Werner Bihlmaier, Jochen Geittner, Rolf Huisgen e Hans-Ulrich ReissigP, The Stereospecificity of Diazomethane Cycloadditions, in Heterocycles, vol. 10, 1978, pp. 147–152, DOI:10.3987/S-1978-01-0147.
- ^ Rolf Huisgen, Wolfgang Scheer e Helmut Huber, Stereospecific Conversion of cis-trans Isomeric Aziridines to Open-Chain Azomethine Ylides, in Journal of the American Chemical Society, vol. 89, n. 7, 1967, pp. 1753–1755, DOI:10.1021/ja00983a052.
- ^ Alexander Dahmen, Helmut Hamberger, Rolf Huisgen e Volker Markowski, Conrotatory ring opening of cyanostilbene oxides to carbonyl ylides, in Journal of the Chemical Society D: Chemical Communications, vol. 0, n. 19, 1971, pp. 1192–1194, DOI:10.1039/C29710001192.
- ^ Vsevolod V. Rostovtsev, Luke G. Green, Valery V. Fokin e K. Barry Sharpless, <2596::AID-ANIE2596>3.0.CO;2-4 A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes, in Angewandte Chemie International Edition, vol. 41, n. 14, 2002, pp. 2596–22599, DOI:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4, PMID 12203546.
- ^ Pierluigi Caramella e K.N. Houk, Geometries of nitrilium betaines. The clarification of apparently anomalous reactions of 1,3-dipoles, in Journal of the American Chemical Society, vol. 98, n. 20, 1976, pp. 6397–6399, DOI:10.1021/ja00436a062.
- ^ Pierluigi Caramella, Ruth W. Gandour, Janet A. Hall, Cynthia G. Deville e K. N. Houk, A derivation of the shapes and energies of the molecular orbitals of 1,3-dipoles. Geometry optimizations of these species by MINDO/2 and MINDO/3, in Journal of the American Chemical Society, vol. 99, n. 2, 1977, pp. 385–392, DOI:10.1021/ja00444a013.
- ^ Albert Padwa, 1,3-Dipolar Cycloaddition Chemistry, General Heterocyclic Chemistry Series, vol. 1, United States of America, Wiley-Interscience, 1983, pp. 141–145, ISBN 978-0-471-08364-1.
Voci correlate modifica
Altri progetti modifica
 Wikimedia Commons contiene immagini o altri file su Cicloaddizione 1,3-dipolare
Wikimedia Commons contiene immagini o altri file su Cicloaddizione 1,3-dipolare