Emangioblastoma
Gli emangioblastomi sono tumori del sistema nervoso centrale originati dal sistema vascolare, generalmente durante la mezza età. A volte questi tumori si verificano in altri siti come il midollo spinale e la retina.[1] Possono essere associati ad altre malattie come la policitemia (aumento della conta delle cellule del sangue), cisti pancreatiche e sindrome di von Hippel-Lindau (sindrome VHL). Gli emangioblastomi sono più comunemente composti da cellule stromali nei piccoli vasi sanguigni e di solito si verificano nel cervelletto, nel tronco encefalico o nel midollo spinale. Sono classificati come tumori di primo grado secondo il sistema di classificazione dell'Organizzazione Mondiale della Sanità (OMS).[2]
| Emangioblastoma | |
|---|---|
 | |
| Malattia rara | |
| Specialità | Oncologia |
| Sede colpita | |
| Classificazione e risorse esterne (EN) | |
| ICD-O | 9161/1 |
| ICD-10 | D48.1 |
| MeSH | D018325 |
| eMedicine | 340994 |
 | |
Presentazione modifica
Complicazioni modifica
Gli emangioblastomi possono causare un numero anormalmente elevato di globuli rossi nel flusso sanguigno a causa della produzione ectopica dell'ormone eritropoietina come una sindrome paraneoplastica.
Patogenesi modifica
Gli emangioblastomi sono composti da cellule endoteliali, periciti e cellule stromali. Nella sindrome VHL il soppressore del tumore Von Hippel-Lindau (pVHL) è disfunzionale, generalmente a causa di mutazione e/o silenziamento genico. In circostanze normali, il pVHL è coinvolto nell'inibizione del fattore 1 α inducibile dall'ipossia (HIF-1α) per degradazione proteasomica mediata dall'ubiquitina. In queste cellule disfunzionali il pVHL non può degradare l'HIF-1α, causandone l'accumulo. L'HIF-1α provoca la produzione del fattore di crescita dell'endotelio vascolare, del fattore B di crescita derivato dalle piastrine, dell'eritropoietina e del fattore di crescita trasformante alfa, che agiscono stimolando la crescita delle cellule all'interno del tumore.[3]
Diagnosi modifica

La diagnosi primaria viene fatta con una tomografia assiale computerizzata (TAC). Su una TAC, l'emangioblastoma si presenta come una regione ben definita, a bassa attenuazione nella fossa cranica posteriore con un nodulo che si accresce sulla parete. A volte sono presenti più lesioni.[1]
Trattamento modifica
Il trattamento dell'emangioblastoma è l'escissione chirurgica del tumore.[4] Sebbene di solito sia semplice da eseguire, in circa il 20% dei pazienti si verificano recidive o più tumori in un sito diverso.[1] Anche la radiochirurgia Gamma Knife e l'LINAC sono state impiegate per trattare con successo la recidiva e controllare la crescita tumorale degli emangioblastomi cerebellari.
Prognosi modifica
L'esito dell'emangioblastoma è molto buono, se è possibile ottenere l'estrazione chirurgica del tumore; l'escissione è possibile nella maggior parte dei casi e il deficit neurologico permanente è raro e può essere evitato del tutto se il tumore viene diagnosticato e trattato in anticipo. Le persone con sindrome VHL hanno una prognosi peggiore rispetto a quelle che hanno tumori sporadici poiché quelle con sindrome VHL di solito hanno più di una lesione.[2]
Epidemiologia modifica
L'emangioblastoma è tra i tumori del sistema nervoso centrale più rari e rappresenta meno del 2%. Gli emangioblastomi di solito si verificano negli adulti, ma i tumori possono comparire nella sindrome VHL in età molto più giovane. Uomini e donne sono approssimativamente allo stesso rischio. Sebbene possano verificarsi in qualsiasi sezione del sistema nervoso centrale, di solito si verificano in entrambi i lati del cervelletto, del tronco encefalico o del midollo spinale.[2][5]
Immagini addizionali modifica
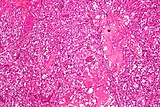
- Micrografie dell'emangioblastoma cerebellare. Colorazione HPS.
-
-


- Emangioblastomi nella malattia di von Hippel-Lindau.
-

-



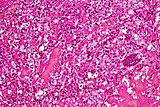
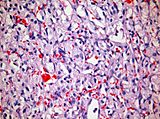
- Istopatologia degli emangioblastomi.
-

-

-

-




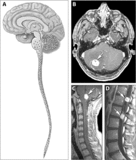
- Risonanza magnetica degli emangioblastomi nella malattia di von Hippel-Lindau.
-

-

-
-
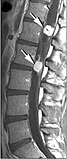
-



.jpg)
Note modifica
- ^ a b c Kenneth W Lindsay, Ian Bone, Robin Callander e J. van Gijn, Neurology and Neurosurgery Illustrated, United States, Churchill Livingstone, 1991, ISBN 0-443-04345-0.
- ^ a b c David N Louis, WHO Classification of Tumors of the Central Nervous System, IARC, 1991, ISBN 92-832-2430-2.
- ^ William G Kaelin, von Hippel–Lindau-associated malignancies: Mechanisms and therapeutic opportunities, in Drug Discovery Today: Disease Mechanisms, vol. 2, n. 2, 2005, pp. 225–231, DOI:10.1016/j.ddmec.2005.04.003.
- ^ Resezione di un emangioblastoma cerebellare
- ^ Richard S, Campello C, Taillandier L, Parker F, Resche F, Haemangioblastoma of the central nervous system in von Hippel-Lindau disease. French VHL Study Group, in J. Intern. Med., vol. 243, n. 6, 1998, pp. 547–53, PMID 9681857.
Voci correlate modifica
Altri progetti modifica
 Wikimedia Commons contiene immagini o altri file su Emangioblastoma
Wikimedia Commons contiene immagini o altri file su Emangioblastoma