Sindrome di Marfan
La sindrome di Marfan è una patologia autosomica dominante (MIM/OMIM 154700) che colpisce il tessuto connettivo[1]. La sindrome si caratterizza per una anomala produzione della proteina fibrillina1 causata da una mutazione del gene FBN1[2] situato nel cromosoma 15[3][4]. Le manifestazioni della sindrome di Marfan interessano gli organi che contengono tessuto connettivo come il sistema scheletrico, gli occhi, il cuore e i vasi sanguigni, i polmoni e le membrane fibrose che ricoprono il cervello e la colonna vertebrale. Il termine deriva dal nome del pediatra francese Antoine Marfan, che per primo descrisse la sindrome nel 1896 in una bambina di cinque anni.
| Sindrome di Marfan | |
|---|---|
| Malattia rara | |
| Cod. esenz. SSN | RN1320 |
| Specialità | genetica clinica |
| Classificazione e risorse esterne (EN) | |
| OMIM | 154700 |
| MeSH | D008382 |
| MedlinePlus | 000418 |
| eMedicine | 946315 e 1258926 |
| GeneReviews | Panoramica |
| Eponimi | |
| Antoine Marfan | |
Epidemiologia
modificaSi stima che un bambino ogni 5000 nati vivi sia affetto dalla sindrome, ma l'incidenza è probabilmente sottostimata per via delle frequenti forme fruste, che non manifestandosi con un fenotipo peculiare possono facilmente sfuggire alla diagnosi. Maschi e femmine sono colpiti in egual misura; non esistono distinzioni di etnia nell'incidenza della patologia. Diversi personaggi storici ne sono stati indicati come probabilmente affetti: Abraham Lincoln, Charles de Gaulle, Niccolò Paganini, il faraone Akhenaton, Charles Maurice de Talleyrand-Périgord, Reinhard Heydrich.
Circa il 75% degli affetti ha almeno un genitore affetto a sua volta, con trasmissione genica di tipo autosomico dominante; tuttavia, nel 25% dei casi, non si riscontrano altri pazienti Marfan nella propria famiglia: in tal caso si tratta quindi di una mutazione genetica de novo a livello degli spermatozoi paterni. Pare che le mutazioni de novo siano più frequenti se l'età del padre del concepito supera i 45 anni.[5]
Segni e sintomi
modificaLo spettro delle manifestazioni della sindrome è molto ampio e diversificato: mentre per alcuni pazienti la diagnosi è immediata e precoce, per quei soggetti invece per i quali solo alcuni dei sintomi sono presenti la diagnosi è difficoltosa. Solo un'indagine genetica (con sensibilità altissima) può in definitiva garantire una diagnosi precisa per queste persone che, paradossalmente, sono quelle più a rischio a causa dell'assenza di attento monitoraggio periodico del loro apparato cardiovascolare.[6]
Apparato locomotore
modificaNelle manifestazioni muscolo-scheletriche, ciò che più colpisce è l'altezza dei pazienti, che sono di solito più alti della media dei coetanei, e spesso sviluppano (soprattutto durante l'adolescenza) segni di magrezza eccessiva (intesa non soltanto come un semplice sottopeso, ma anche come un habitus in generale eccessivamente affusolato e dinoccolato: questo per via soprattutto di arti particolarmente lunghi e sottili rispetto al tronco, aspetto detto "dolicostenomelia": si tratta del cosiddetto habitus marfanoide). Uno studio condotto in Corea del Sud mostra che il 50° percentile dell'altezza degli adulti Marfan supera il 97° percentile della popolazione generale, mentre il 50° percentile del peso dei Marfan supera di poco il 75° percentile della popolazione sana nei maschi ed è compreso tra il 50° e il 75° percentile della popolazione sana nelle femmine; ciò indica un peso corporeo tendenzialmente basso in rapporto ad un'altezza che risulta quasi sempre sopra la norma.[7] La muscolatura è spesso ridotta con habitus ectomorfico e difficoltà ad acquisire massa muscolare, anche se senza ipoplasia grave - più seria è in genere la carenza muscolare osservabile nella sindrome di Beals. Può essere presente una ridotta resistenza allo sforzo fisico.
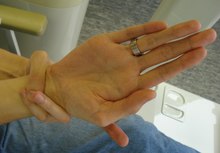
Un'altra caratteristica è la lunghezza, iperestensibilità e forma affusolata delle dita, che viene chiamata "aracnodattilia" ed è riconoscibile dal segno del pollice o segno di Steinberg (l'intera unghia del pollice flesso verso il palmo della mano oltrepassa il bordo ulnare) e dal segno del polso o segno di Walker-Murdoch (l'estremità del mignolo e del pollice di una stessa mano riescono a sovrapporsi facendo il giro attorno al polso dell'altro braccio); l'aracnodattilia, che tende a causare una ipermobilità delle dita, è legata alla lassità generalizzata dei legamenti[6]. Comune in malattie come la sindrome di Marfan e la malattia ossea di Paget, ma molto rara in pazienti altrimenti sani, è la protrusione dell'acetabolo, che spesso peggiora con l'età. Tale protrusione, insieme all'ectasia durale e in generale ad una predisposizione a sviluppare stanchezza cronica, porta molti affetti dalla sindrome a sviluppare dolore cronico e mancanza di energie nel corso degli anni; si riscontra anche una degenerazione ossea, con osteopenia e in alcuni casi osteoporosi ad esordio anticipato rispetto alla popolazione generale.
Molto frequenti e peculiari sono le deformità del petto, dovute non tanto a una deformazione dello sterno, quanto piuttosto ad un sovrasviluppo e una conseguente estroflessione o introflessione delle costole; tali anomalie si chiamano rispettivamente petto carenato e petto escavato: esse sono spesso molto evidenti, tuttavia solo nei casi più eclatanti impediscono il raggiungimento della massima capacità polmonare in inspirazione (e sono quindi da risolvere con un intervento chirurgico).[8] Anche la coesistenza di cifosi e di scoliosi superiore a 20°, le quali tendono a peggiorare con l'età, è ricorrente nella sindrome; per una scoliosi che supera i 40°-45° è spesso consigliato un intervento chirurgico correttivo. Nei pazienti Marfan si riscontra inoltre, a livello podologico, un'aumentata incidenza di piede piatto e di deformazioni del retropiede, con tallone flesso verso l'esterno; anche l'alluce valgo non è raro. Altra caratteristica ortopedica piuttosto frequente nella sindrome è il genu recurvatum, cioè ginocchia spostate all'indietro rispetto al piano verticale dell'arto inferiore.
Sul piano odontoiatrico e gnatologico sono molto frequenti la micrognazia, la retrognazia, il palato ogivale (palato stretto) con conseguente malocclusione (spesso di tipo 2) e l'ipoplasia mascellare. Tutto ciò aumenta la probabilità di avere una dentatura anomala, con denti accavallati, specie nell'arcata superiore. Queste anormalità del cavo orale, unite alla debolezza intrinseca dei muscoli delle alte vie aeree, sono responsabili di una maggior incidenza di apnea notturna di tipo ostruttivo nei pazienti Marfan rispetto alla popolazione generale.[9]
Apparato cardiovascolare
modificaL'apparato cardiovascolare comprende delle alterazioni talora molto gravi: la porzione ascendente dell'aorta tende a dilatarsi, con conseguente indebolimento della tonaca media arteriosa che con un circolo vizioso regge con sempre più difficoltà la pressione sanguigna portando la dilatazione ad aumentare progressivamente, diventando aneurismatica dopo un'iniziale fase di ectasia. L'occorrenza di una dissecazione dell'aorta ascendente, molto più frequente in caso di aneurisma conclamato ma possibile anche con diametri aortici nei limiti della norma, è tipica della sindrome e tende ad avvenire entro la mezza età (sono interessati soprattutto il bulbo aortico e la radice aortica). È meno frequente, ma non raro, anche lo sviluppo di aneurismi a livello dell'arco aortico o dell'aorta discendente, compresa l'aorta addominale. Mentre un paziente diagnosticato sarà seguito durante tutta l'evoluzione della malattia e quindi monitorato con ecocardiogramma per le sue eventuali modifiche delle misure aortiche (e, nel caso, operato prima che si arrivi alla dissecazione), le persone che non ricevono una diagnosi rischiano di essere colpite improvvisamente dalla patologia aortica con conseguenze spesso drammatiche.[6] Il 70% degli affetti da sindrome di Marfan arriva a presentare un diametro aortico anormale già entro i 20 anni di vita, l'80% entro i 40 anni, il 35% già entro i primi 5 anni di vita.[10]
Il diametro della radice aortica è direttamente proporzionale al rischio di dissezione aortica e di rottura: l'aorta dei pazienti Marfan ha una fragilità intrinseca e quindi è a rischio di dissezione anche con diametri entro i limiti della norma, tuttavia il rischio di eventi aortici, in caso di adeguata terapia betabloccante e di astensione da sforzi fisici intensi, resta ben sotto l'1% annuo con diametri dell'aorta ascendente inferiori a 50 mm, mentre cresce esponenzialmente oltre questa soglia, diventando circa del 5% annuo in caso di diametro attorno ai 60 mm.[11]
Da ricordare l'insufficienza della valvola mitrale (presente nel 50-60% dei pazienti Marfan) e della tricuspide; talora queste valvulopatie sono dovute al prolasso dei lembi, talora alla rottura delle corde tendinee.[6] Anche la valvola aortica è spesso insufficiente; sono frequenti i rigurgiti mitralici, aortici e tricuspidalici. Gravi valvulopatie possono condurre all'insufficienza cardiaca congestizia. Il prolasso della mitrale spesso progredisce con l'età, portando nei casi più severi alla deiscenza della valvola.[12] Questa anomalia valvolare può essere corretta mediante un intervento chirurgico (effettuabile con la sostituzione della valvola biologica con una meccanica, oppure con il mantenimento di valvole biologiche), a seguito del quale il tasso di sopravvivenza è ottimo e il rischio aritmogenico a lungo termine è basso.[13]
La presenza di aritmie ventricolari importanti (tra cui tachicardia ventricolare sostenuta o non sostenuta, così come frequenti battiti ectopici ventricolari), sia in assenza sia in comorbidità con valvulopatie significative, espone ad un maggior rischio di morte cardiaca improvvisa i pazienti Marfan, incrementandone la mortalità per arresto cardiaco e per tutte le cause rispetto ai Marfan con più blande o assenti manifestazioni aritmiche.[14] I pazienti Marfan sono ad alto rischio di sviluppare un'endocardite infettiva con danni soprattutto a carico della valvola mitrale; per ridurre questa possibilità si consiglia una terapia antibiotica a scopo preventivo prima di essere sottoposti ad interventi chirurgici di ogni sorta, soprattutto quelli ortodontici.
Occhio
modifica
Le manifestazioni oculari sono fondamentalmente caratterizzate dal dislocamento del cristallino (chiamato anche ectopia lentis) conseguente a una sublussazione dello stesso e dalla sferofachia[15], spesso presente nelle forme diagnosticate in età infantile e da problemi della retina, come il distacco.[6] La dislocazione del cristallino, in particolare, è un segno molto peculiare della sindrome: può avvenire a qualsiasi età e può essere monolaterale o bilaterale. Quando insorge in età avanzata tende ad essere più grave, bilaterale e dall'evoluzione particolarmente rapida. Le suddette anomalie oculari causano molto spesso vizi di rifrazione: tra di essi il più frequente è una miopia spesso molto forte, a esordio spesso infantile, ma si possono riscontrare anche presbiopia e astigmatismo.[16] Si può avere ipermetropia nei casi più gravi di sublussazione del cristallino. Frequente è anche lo strabismo, sia esotropico sia exotropico, ad esordio generalmente infantile.[17] Anche il coloboma dell'iride è più frequente in caso di sindrome di Marfan. La sindrome espone ad un rischio abbastanza alto di sviluppare cataratta ben prima della vecchiaia e glaucoma ad ogni età.
Altre manifestazioni
modificaUn altro segno tipico, quasi patognomonico della sindrome è l'ectasia durale, che si presenta in oltre la metà dei pazienti Marfan nel corso della loro vita e che consiste nella progressiva dilatazione del rivestimento di dura madre attorno alla colonna vertebrale; in genere avviene a livello lombosacrale, laddove la pressione esercitata dal liquido cefalorachidiano sui tessuti che lo contengono è più elevata. Nelle fasi iniziali l'ectasia durale tende ad essere asintomatica, ma quando si aggrava tende a causare dolori lombosacrali, cefalea, astenia, debolezza e diminuita sensibilità agli arti inferiori, dolori rettali e genitali, incontinenza urinaria, ritenzione urinaria; i sintomi tendono a peggiorare stando in posizione eretta. Per la diagnosi di questa alterazione è necessaria la risonanza magnetica nucleare o la tomografia computerizzata.
A livello polmonare, oltre alle eventuali difficoltà respiratorie dovute a forme gravi delle deformazioni toraciche succitate, si riscontra pneumotorace con aumentata incidenza, specie in soggetti giovani: molteplici eventi di pneumotorace spontaneo sono possibili nei pazienti Marfan ma sono molto rari in soggetti altrimenti sani. Anche l'enfisema pur senza fattori di rischio associati, come il tabagismo, è piuttosto frequente tra gli affetti dalla sindrome, con alterazioni all'auscultazione soprattutto a livello degli apici polmonari. Solo il 37% degli affetti ha una normale funzione polmonare: il 19% ha un pattern restrittivo e il 44% ha addirittura un pattern di ostruzione respiratoria.[18]
Si riscontrano molto spesso, a livello cutaneo, strie cutanee agli arti che compaiono in epoca pre-adolescenziale e possono non essere correlate a grandi cambiamenti di statura o di peso corporeo: tendono a localizzarsi anche in sedi atipiche, come gli arti superiori e le spalle[19]; inizialmente rosse, progressivamente diventano bianche e meno antiestetiche, ma quasi sempre cronicizzano.
Circa il 29% degli individui affetti dalla sindrome presenta alterazioni significative della coagulazione: alterazioni nella struttura delle piastrine possono portare a eccessivi sanguinamenti, analoghi a quelli visibili nella malattia di von Willebrand o in altre trombocitopatie congenite.[20] È stata dimostrata una proporzionalità diretta tra concentrazione ematica di D-dimero e ritmo naturale di progressione della dilatazione aortica in pazienti Marfan adulti, così come una proporzionalità inversa tra grado di attività del fattore VIII di coagulazione e ritmo di dilatazione aortica. Con la sindrome di Marfan il fattore di von Willebrand e lo stesso fattore VIII di coagulazione presentano un'attività mediamente minore che nella popolazione generale, mentre il tempo di tromboplastina parziale (PTT) in genere non risulta alterato.[21]
La generale lassità del tessuto connettivo favorisce la formazione di ernie, in particolare di ernie inguinali, le quali sarebbero presenti in circa il 32% dei pazienti Marfan di oltre 26 anni di età che si sottopongono ad intervento di sostituzione dell'aorta ascendente.[22] Anche lo sviluppo di ernia iatale, di ernia ombelicale - ancora più frequente nella sindrome di Ehlers-Danlos di tipo vascolare - e di malattia da reflusso gastroesofageo (solo alcune volte correlata alla presenza di un'ernia iatale) sono più frequenti nella popolazione Marfan che nella popolazione sana.
Sindrome di Marfan neonatale
modificaCirca il 5-10% delle sindromi di Marfan si manifesta con sintomi seri e rapidamente ingravescenti sin dalla nascita: si tratta della cosiddetta sindrome di Marfan neonatale (che non va confusa con la sindrome di Marfan classica se la diagnosi di quest'ultima avviene in epoca neonatale). Il decorso è molto più severo che nella sindrome di Marfan classica, e si osservano anomalie craniofacciali e oculari fin dalla nascita, tra cui iridodonesi, megalocornea, ectopia lentis, padiglione auricolare accartocciato - similmente a quanto si osserva nella assai meno pericolosa sindrome di Beals -, cute in eccesso a conferire un aspetto "senile" al volto del bambino affetto; si osservano anche molteplici contratture a livello delle articolazioni, enfisema polmonare ingravescente e frequenti vizi valvolari molto gravi, tra cui il prolasso della valvola mitrale e della valvola tricuspide; l'aorta ascendente tende a dilatarsi fin dai primi mesi di vita ed è soggetta a rotture degli aneurismi fin dalla prima infanzia. L'interessamento polmonare è serio, a causa di una grave insufficienza cardiaca: si osservano arterie polmonari dilatate in un quadro di forte ipertensione polmonare. Si riscontrano di frequente anche petto carenato, aracnodattilia e dolicostenomelia.[23] Similmente alla sindrome di Marfan classica, la variante neonatale è conseguenza di una mutazione a carico del gene FBN1;[24] la prognosi della forma neonatale della malattia è molto severa: il 95% dei malati non supera l'anno di vita, con una mediana di circa 5 mesi; l'aspettativa di vita media è però di 16 mesi, a testimonianza di come ci siano alcuni outlier che possono sopravvivere oltre i 4-11 anni, raggiungendo l'adolescenza o addirittura la prima età adulta.[25]
Diagnosi
modificaCriteri diagnostici di Ghent
modificaNel 2010 la nosologia di Ghent è stata aggiornata rispetto alla precedente versione pubblicata nel 1996; i nuovi criteri permettono di diagnosticare la sindrome di Marfan su base clinica, senza test genetico, con elevata sensibilità e specificità. La diagnosi avviene:
In assenza di storia familiare di sindrome di Marfan:
- con uno Z-score della radice aortica maggiore o uguale a 2,00 ED ectopia lentis, oppure
- con uno Z-score della radice aortica maggiore o uguale a 2,00 E una mutazione del gene FBN1, oppure
- con uno Z-score della radice aortica maggiore o uguale a 2,00 E uno score sistemico di 7 o più punti, oppure
- con ectopia lentis E una mutazione del gene FBN1 in presenza di patologia aortica documentata.
In presenza di storia familiare di sindrome di Marfan:
- Ectopia lentis, oppure
- Score sistemico di 7 o più punti, oppure
- Z-score della radice aortica maggiore o uguale a 2,00.
Lo score sistemico in questione è così calcolabile:
- Segno del pollice E segno del polso: 3 punti (segno del pollice O segno del polso: 1 punto).
- Petto carenato: 2 punti (petto escavato o altra asimmetria ossea toracica: 1 punto).
- Deformità del retropiede a livello di astragalo e/o calcagno: 2 punti (piede piatto: 1 punto).
- Ectasia durale: 2 punti.
- Protrusio acetabuli: 1 punto.
- Pneumotorace spontaneo: 2 punti.
- Rapporto tra segmento superiore e segmento inferiore del corpo inferiore a 0,87 E rapporto tra ampiezza delle braccia e altezza superiore a 1,05 E assenza di scoliosi di grado severo: 1 punto.
- Scoliosi e/o cifosi toraco-lombare: 1 punto.
- Estensione del gomito in abduzione inferiore a 170 gradi: 1 punto.
- Presenza di almeno 3 delle seguenti 5 anomalie craniofacciali: enoftalmo, dolicocefalia, rime palpebrali piegate verso il basso, ipoplasia malare, retrognazia: 1 punto.
- Smagliature: 1 punto.
- Miopia superiore a 3 diottrie: 1 punto.
- Prolasso della valvola mitrale: 1 punto.
Diagnosi differenziale
modificaLa sindrome di Marfan entra in diagnosi differenziale con altre connettivopatie e altre malattie genetiche:
- Sindrome di Beals, che presenta aracnodattilia, cifoscoliosi ed ectasia dell'aorta ascendente ma presenta contratture articolari indolori congenite, con camptodattilia e ipotonia generalizzata medio-grave, che sono infrequenti nella sindrome di Marfan.
- Fenotipo MASS, che condivide le anomalie scheletriche, articolari e delle valvole cardiache della sindrome di Marfan ma non presenta una patologia aortica che progredisce con la stessa severità.
- Sindrome di Stickler, che può dare anomalie craniofacciali, distacco di retina, grave miopia e altri sintomi presenti nella sindrome di Marfan, ma in genere dà un importante coinvolgimento auricolare con progressiva sordità, cosa che non avviene nella sindrome di Marfan.
- Sindrome di Shprintzen-Goldberg, malattia rarissima e diagnosticata solo negli ultimi decenni; comporta anomalie facciali più gravi di quelle della sindrome di Marfan e, soprattutto, molto spesso è accompagnata da ritardo mentale.
- Sindrome di Ehlers-Danlos, che provoca un'ipermobilità articolare anche superiore a quella della sindrome di Marfan, alterazioni cutanee e altri sintomi tipici dei difetti congeniti del tessuto connettivo; la forma vascolare della sindrome di Ehlers-Danlos inoltre può dare aneurismi dissecanti non solo all'aorta, come nel caso della sindrome di Marfan, ma anche a molte altre arterie che nel caso della sindrome di Marfan sono alterate solo di rado.
- Omocisteinuria, che causa difetti scheletrici analoghi a quelli della sindrome di Marfan (soprattutto petto carenato, cifoscoliosi, piede varo) e difetti oculari particolari come l'ectopia lentis e la cataratta, ma molto spesso comporta un ritardo mentale che invece, nei soggetti Marfan, ha la stessa prevalenza che ha nella popolazione generale.
- Neoplasia multipla endocrina di tipo 2B, che conferisce un aspetto "marfanoide" agli individui affetti (alta statura, magrezza eccessiva, arti affusolati, leggera ipoplasia muscolare).
- Sindrome di Loeys-Dietz, che comporta anch'essa ectasia durale ma si distingue dalla sindrome di Marfan per frequente ugola bifida, altre anomalie al palato, anomalie nella cicatrizzazione delle ferite ed ipertelorismo oculare.
- Sindrome di Klinefelter, che come la sindrome di Marfan determina arti molto lunghi rispetto al tronco e alta statura; pur non avendo nel complesso molti segni in comune con la sindrome di Marfan, trattandosi di una malattia relativamente comune, è spesso erroneamente diagnosticata al posto della sindrome di Marfan stessa.
Terapia
modificaL'unica strategia praticata per rallentare l'indebolimento e l'allargamento dell'aorta è l'impiego di farmaci per ridurre la pressione sanguigna. Di solito vengono prescritti betabloccanti ma ultimamente la pratica clinica sembra evidenziare un maggior beneficio con i sartani (losartan, telmisartan, eccetera) e con gli ACE inibitori. Studi sono in corso per accertarne l'effettiva efficacia. In particolare è emerso, durante uno studio su un ridotto numero di pazienti con sindrome di Marfan, che il perindopril risulterebbe particolarmente efficace. Lo studio condotto presso il Baker Heart Institute a Melbourne ha evidenziato una sensibile diminuzione del diametro aortico, della rigidità dell'aorta ed un conseguente aumento di elasticità con minor rischio di rottura.[26]
Sono stati pubblicati i risultati di uno studio "piccolo" ma ben organizzato che evidenziano per il perindopril, il verapamil e l'atenololo una riduzione della pressione sistolica periferica e centrale significativa, ma solo i beta bloccanti, rallentando la frequenza cardiaca e ritardando l'onda aortica, possono avere un ruolo costante nella riduzione dell'espansione ritardata nell'arco e nell'aorta addominale[27]. Ricerche hanno dimostrato che i sartani, in particolare il losartan, tendono a rallentare in modo significativo la dilatazione dell'aorta ascendente[28], ma successivi trial non hanno confermato questa osservazione.[29]
La terapia chirurgica (che prevede una sternotomia e successivamente una sostituzione della porzione aortica interessata dall'aneurisma, durante la quale ci si avvale di macchinari per la circolazione extracorporea) è rivolta ad individuare il giusto "timing chirurgico" per la correzione della dilatazione aortica prima che si raggiungano livelli pericolosi, che possono portare alla dissezione dell'aorta acuta. Studi hanno evidenziato che un diametro aortico di 50 mm possa considerarsi il cut off per intervenire in tempi relativamente sicuri.[30]. Altri studi più recenti, alla luce di un diminuito rischio intraoperatorio per le sostituzioni della radice aortica, considerano invece i 45 mm come limite di diametro aortico oltre il quale è consigliabile l'operazione. Molto spesso durante questi interventi si ricorre all'impianto di una protesi non-organica in Dacron che va a sostituire il tratto dell'aorta interessato dalla dilatazione aneurismatica, talvolta secondo l'intervento di Bentall - eseguito per la prima volta nel 1968 e più diffusamente da fine anni Settanta[31] - che prevede la sostituzione della valvola aortica con una valvola meccanica, talvolta secondo l'intervento di David che implica la conservazione della valvola aortica naturale - spesso non praticabile in caso di insufficienza valvolare ingravescente. Dopo il primo intervento vi è necessità di seguire una terapia anticoagulante orale per tutta la vita e sussiste comunque un aumentato rischio tromboembolico, mentre la seconda tecnica operatoria non necessita di successiva terapia anticoagulante ma espone a un maggior rischio di dover essere sottoposti ad un nuovo intervento (tale rischio è stimato essere dell'1,3% annuo).
La mortalità peri-operatoria è molto più alta (anche oltre il 10% entro 30 giorni dall'intervento) in caso di intervento in emergenza, con dissezione acuta in corso, che nel caso di un intervento elettivo (senza il contesto emergenziale della dissezione), per il quale la mortalità a 30 giorni scende all'1,5% circa.[32]
Prognosi
modificaFino a inizio anni '70 l'aspettativa di vita mediana per i malati Marfan era di circa 40 anni per gli uomini e di pochi anni più alta per le donne; la morte poteva avvenire anche in età molto giovane, essenzialmente per dissecazione aortica e conseguente rottura del vaso, nonché per insufficienza cardiaca legata all'aggravamento delle condizioni di prolasso mitralico ed insufficienza aortica. Sono stati compiuti notevoli miglioramenti in questo senso; grazie ad un adeguato stile di vita (evitando sport pericolosi per le pareti aortiche e che innalzano molto la pressione arteriosa), la possibilità di interventi chirurgici su aorta, cuore ed altre parti dell'organismo, metodi diagnostici e di screening più efficienti e medicinali che aiutano a limitare e a posticipare le principali complicanze della malattia, la speranza di vita - stando a molteplici studi - non differisce molto da quella della popolazione generale.
Altre ricerche vedono ancora un'ampia differenza nella mortalità dei pazienti Marfan rispetto ai soggetti sani: uno studio norvegese conclusosi nel 2015 su una coorte di pazienti con sindrome di Marfan ha rilevato un'aspettativa di vita mediana pari a 63 anni per i maschi e a 73 anni per le femmine, con una mortalità per tutte le cause rispetto alla popolazione generale dello stesso sesso più alta di 8 volte negli uomini e di 4 volte nelle donne Marfan: pur essendo un dato in aumento rispetto a uno studio danese condotto negli anni '90, che vedeva un'età mediana di morte di 58 anni per i maschi Marfan e di 63 anni per le donne affette, esso mostra una differenza significativa rispetto alla sopravvivenza media della popolazione generale norvegese. I problemi cardiovascolari, in particolare a livello dell'aorta (dissezioni anche decenni dopo gli interventi chirurgici di sostituzione di parti dell'arteria), sembrano essere tuttora la causa più frequente di decesso tra i pazienti Marfan.[33]
Oltre a determinare una mortalità perioperatoria molto superiore, un intervento emergenziale di riparazione dell'aorta nel corso di una dissezione acuta sembra essere correlato ad tasso di sopravvivenza più basso anche sul lungo periodo: uno studio ha calcolato una sopravvivenza a 20 anni superiore al 70% nel caso di sostituzione della radice aortica a scopo preventivo, ma inferiore al 50% se lo stesso intervento è effettuato in condizioni di urgenza o di emergenza (dissecazione incipiente o acuta).[32]
Celebrità affette dalla sindrome
modifica- John Tavener (1944-2013), compositore britannico.[34]
- Isaiah Austin (1993-), cestista statunitense.[35]
- Jonathan Jeanne (1997-), cestista francese.[36]
- Dominique de Roux (1935-1977), scrittore francese.
- Javier Botet (1977-), attore spagnolo.
- Troye Sivan (1995-), cantautore australiano.
- Peter Mayhew (1944-2019), attore britannico.[37]
- Bradford Cox (1982-), cantante statunitense.[38]
- Luciano Vendemini (1952-1977), cestista italiano. [39]
- Vincent Schiavelli (1948-2005), attore italo-americano.
- Niccolò Paganini (1782-1840) è stato un violinista, violista, chitarrista e compositore italiano
Nell'arte
modificaMO (2007) è un film del regista statunitense Brian Scott Lederman, con protagonista Erik Per Sullivan, in cui si parla di un teenager che soffre della sindrome di Marfan. Il film è ispirato alla vita di Matthew Benjamin Lederman, fratello del regista, deceduto in giovane età a causa della sindrome.
L'attore spagnolo Javier Botet è noto per aver interpretato personaggi in film horror caratterizzati da deformità fisiche ottenibili senza effetti speciali solo grazie alle peculiarità specifiche della sindrome di Marfan. Tra questi, Slender Man e The Conjuring 2.
Note
modifica- ^ 17.4.4 Malattie del tessuto connettivo | SPREAD Live 2011
- ^ HUGO Gene Nomenclature Committee (HGNC), Symbol Report: FBN1, su genenames.org. URL consultato il 26 lug 2017 (archiviato dall'url originale il 14 marzo 2018).
- ^ Luigi Chiariello, "Sindrome di marfan, percorso diagnostico terapeutico assistenziale" (PDF), Fondazione Policlinico Tor Vergata, dicembre 2012. URL consultato il 26 lug 2017 (archiviato dall'url originale il 14 marzo 2018).
- ^ (EN) Jane Kelly - updated : 14/07/2015, Marfan Syndrome; MFS, su omim.org, 6 giugno 2017. URL consultato il 26 luglio 2017.
- ^ What causes Marfan syndrome?, su childrensnational.org.
- ^ a b c d e Sindrome di Marfan, su atlantemedicina.wordpress.com.
- ^ Disease-specific Growth Charts of Marfan Syndrome Patients in Korea
- ^ Pulmonary function in the Marfan syndrome
- ^ High prevalence of obstructive sleep apnea in Marfan's syndrome
- ^ Relation of aortic root dilatation and age in Marfan's syndrome
- ^ Aortic Event Rate in the Marfan Population
- ^ Zipes, Libby Bonow Braunwald (2005). Braunwald's Heart Disease ~ A Textbook of Cardiovascular Medicine, Seventh Edition. United States of America: Elseview Saunders. pag. 1894
- ^ Results of modern mitral valve repair in patients with Marfan syndrome
- ^ Observational Cohort Study of Ventricular Arrhythmia in Adults with Marfan Syndrome Caused by FBN1 Mutations
- ^ Giuseppe Canepa, Sindromi dismorfiche e malattie costituzionali dello scheletro, Padova, Piccin, 1996, pp. 1947.
- ^ About Marfan Syndrome (in inglese)
- ^ Marfan Syndrome su omim.org (inglese)
- ^ Pulmonary involvement in patients with Marfan Syndrome
- ^ A case-control study of cutaneous signs in adult patients with Marfan disease: diagnostic value of striae
- ^ Coagulation Disorders in Marfan Syndrome (PDF)
- ^ Hemostatic abnormalities in adult patients with Marfan syndrome
- ^ Collagenopathies—Implications for Abdominal Wall Reconstruction: A Systematic Review
- ^ Neonatal Marfan syndrome
- ^ A Recurring FBN1 Gene Mutation in Neonatal Marfan Syndrome
- ^ A Case of Neonatal Marfan Syndrome: A Management Conundrum and the Role of a Multidisciplinary Team
- ^ Effect of perindopril on large artery stiffness and aortic root diameter in patients with Marfan syndrome: a randomized controlled trial
- ^ Effects of atenolol, perindopril and verapamil on haemodynamic and vascular function in Marfan syndrome – a randomised, double-blind, crossover trial
- ^ Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial
- ^ Losartan Versus Atenolol for Prevention of Aortic Dilation in Patients With Marfan Syndrome
- ^ Cardiovascular SurgeryAortic Event Rate in the Marfan PopulationA Cohort Stud
- ^ The Button Bentall procedure
- ^ a b Replacement of the Aortic Root in Patients with Marfan's Syndrome
- ^ Survival, causes of death, and cardiovascular events in patients with Marfan syndrome
- ^ Morto John Tavener
- ^ Isaiah Austin has Marfan Syndrome (in lingua inglese)
- ^ Jeanne souffre de la mème maladie qu'Isaiah Austin (in francese)
- ^ Cenno alla sindrome di Marfan nella biografia di Mayhew su infarawaygalaxy.com
- ^ Cenno alla sindrome di Marfan nella biografia di Cox su AllMusic.com
- ^ Cold case: Luciano Vendemini, cronaca di una morte annunciata di Massimo Renella (18-fine), su superbasket.it, Superbasket, 17 febbraio 2018.
Altri progetti
modifica Wikimedia Commons contiene immagini o altri file sulla sindrome di Marfan
Wikimedia Commons contiene immagini o altri file sulla sindrome di Marfan
Collegamenti esterni
modifica- OMIM, su ncbi.nlm.nih.gov.
- Marfan Life Forum, su marfanlife.net. URL consultato il 14 gennaio 2009 (archiviato dall'url originale il 12 febbraio 2009).
- (EN) Marfan Syndrome Forum, su forum.marfan-forum.org.uk. URL consultato il 27 dicembre 2022 (archiviato dall'url originale il 24 dicembre 2012).
- Marfan Friends World, su marfan-friends-world.org.uk. URL consultato il 30 giugno 2010 (archiviato dall'url originale il 30 marzo 2010).
- Marfan Clinic, su marfanclinic.it.
- Marfan Bologna e Regione Emilia Romagna, su aosp.bo.it.
| Controllo di autorità | GND (DE) 4168872-7 |
|---|